
What's New
- A case study of Exemptia in Hidradenitis Suppurativa published in IJDVL Read More..
- A case of relapsing polychondritis with laryngeal stenosis successfully controlled with Exemptia’ accepted in Indian Journal of Rheumatology.
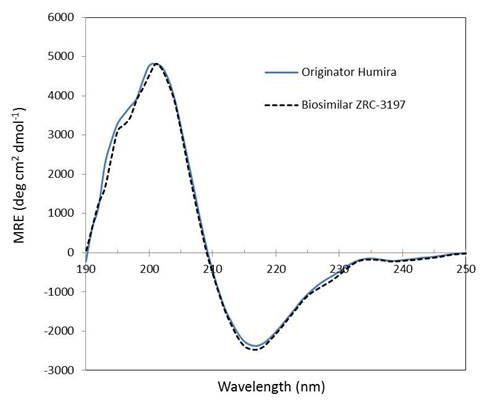
Exemptia contains adalimumab which is biosimilar of originator’s adalimumab. It is an exact fingerprint match of originator’s adalimumab. Biosimilars are defined as ‘similar’ or ‘highly similar’ to the reference medicinal products (originator products) following the EMA (European Medicines Agency) and FDA (Food and Drug Administration, USA) regulatory guidelines. Biosimilars having the similar level of efficacy and safety compared to that of the originator products provide additional advantage to patients in terms of affordability and lost-cost of therapy, while expanding the patient access to therapies. Despite of its complex molecular nature, a target-directed approach for development has been proven highly successful to generate monoclonal antibodies similar to the originator products at the level of individual quality attributes with same functional properties.
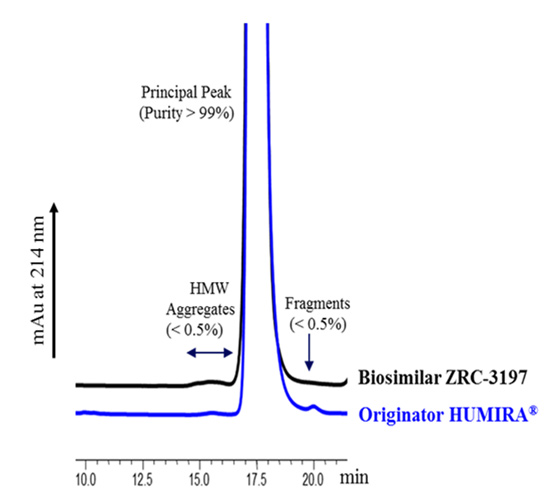
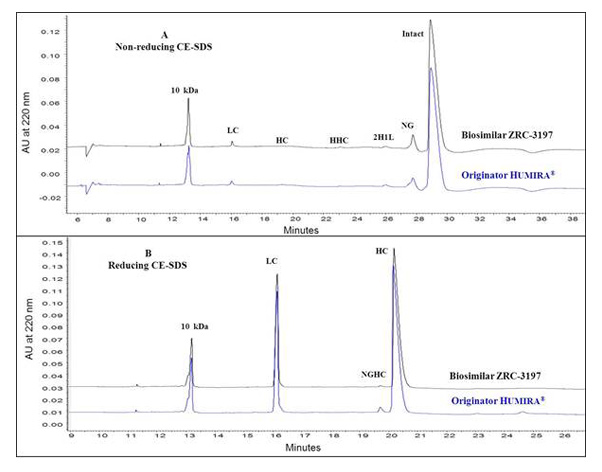
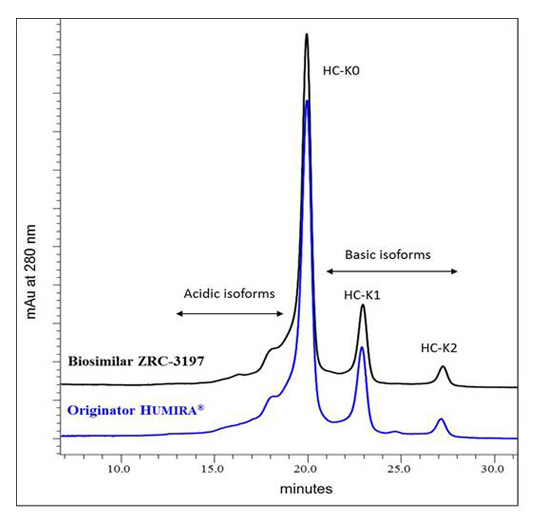
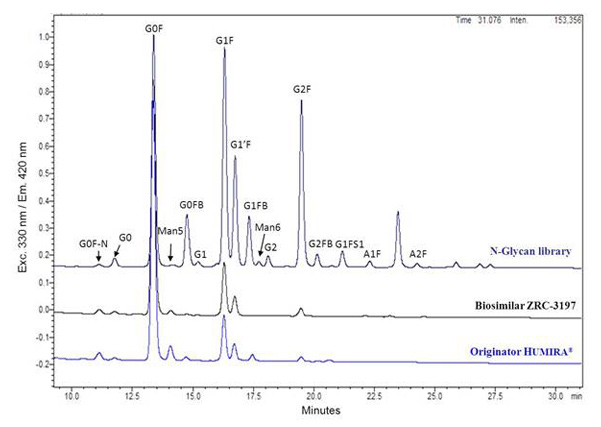
Exemptia, termed as ZRC-3197, has been developed indigenously by Zydus-Cadila (Cadila Healthcare Ltd.), as biosimilar medicinal product of originator HUMIRA®. Originator HUMIRA®, a TNF blocker, is used for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease and plaque psoriasis. Adalimumab, which serves the active substance of originator HUMIRA® is a fully human monoclonal IgG1 antibody produced recombinantly in genetically engineered Chinese Hamster Ovary (CHO) cells. Adalimumab is composed of 1330 amino acids with a molecular weight of about 148 kDa. Physicochemical and functional characterization of Exemptia and HUMIRA®, on a comparative basis to demonstrates biosimilarity between both.
Adalimumab binds specifically to TNF-alpha and blocks its interaction with the p55 and p75 cell surface TNF receptors. Upon binding to human TNF, Adalimumab either neutralizes the biological function of TNF by blocking its interaction with the cell surface TNF-receptors or modulates biological responses involving changes in leukocyte migration of adhesion molecules (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 0.1-0.2 nM). TNF plays a central role in initiation, maintenance and progression of several inflammatory diseases and treatment with Adalimumab improves the signs and symptoms of these TNF associated diseases in patients.

ExemptiaTM should be administered subcutaneously.
As per the study done by M H Weisman et al 1, responses to adalimumab were rapid, with 22.2% patients achieving an ACR20 response within 24 hours of dosing.
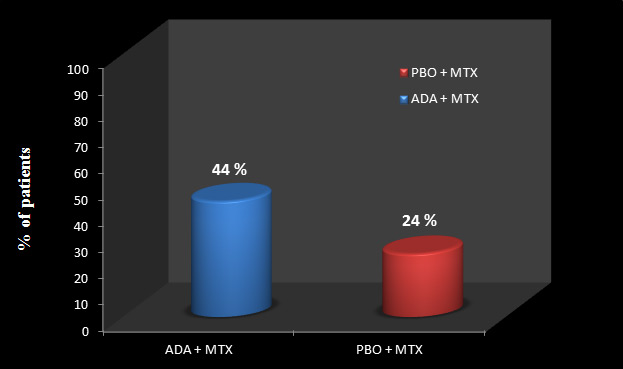
In OPTIMA trial, 2 treatment arms were compared. One arm was given ADA+ MTX and other arm was given placebo (PBO) + MTX. In ADA+ MTX group, 44% of patients achieved stable LDA (DAS28; based on number of swollen and tender joints, C-reactive protein concentration, and patients’ global assessment of disease activity of less than 3.2 at weeks 22 and 26) while only 24% of patients could achieve stable LDA in PBO + MTX group.
Figure 1: Achievement of stable low disease activity
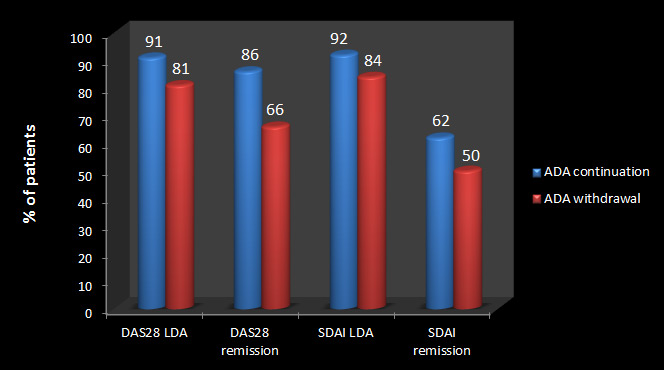
In OPTIMA trial, 44% of patients in ADA+ MTX group who achieved LDA, were again randomized into 2 groups- one is ADA continuation group (received ADA+ MTX, n= 105) and other one was ADA withdrawal group (received only MTX, n= 102). These treatments were given for another 52 weeks and results were compared.
Adalimumab-withdrawal was associated with small and clinically insignificant mean increases in disease activity from week 26 to 78, which were not identified for adalimumab-continuation. Nevertheless, most patients who withdrew adalimumab sustained their clinical responses up to week 78.
Slightly higher proportion of patients who continued, rather than withdrew, adalimumab maintained DAS28 states at week 78 for both DAS28 of less than 3•2 (91% vs.81%) and DAS28 of less than 2•6 (86% vs.66%) [See figure 2].
Same proportions of patients in both groups achieved SDAI low disease activity [92%]for adalimumab-continuation vs. [84%]for adalimumab-withdrawal; p=0•0659) or remission [62%]vs. [50%]; p=0•0988) at week 78 [See figure 2].
The functional improvements identified at week 26 were maintained with adalimumab-withdrawal and were much the same as those identified with adalimumab-continuation.
OPTIMA trial suggests that in most patients with early (the good response will be maintained clinically, functionally, and structurally even when adalimumab is withdrawn. This finding suggests that a short course of an induction therapy using a biological agent in combination with methotrexate might be sufficient to allow maintenance of low disease activity or remission upon continuation of
methotrexate only. This outcome has great implications for care of patients and economic
aspects of treatment of rheumatoid arthritis with expensive biological agents.
There are various studies conducted globally which prove that adalimumab plays a vital role in inhibiting/ minimizing radiographic progression and thus help in preventing joint damage.
Mentioned below are few of those:
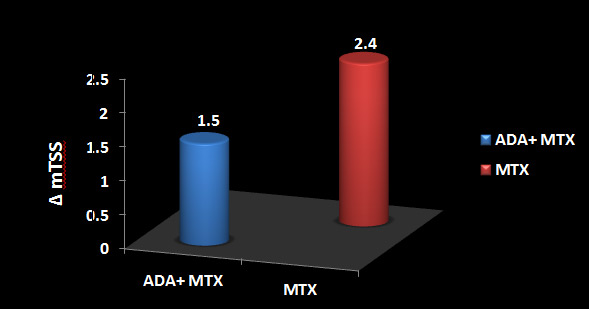
Adalimumab+ MTX significantly inhibited radiographic progression at week 26 versus MTX alone (ΔmTSS, 1.5±6.1 vs. 2.4±3.2, respectively; p<0.001)
(See Figure 1).
Significantly more patients in the adalimumab+ MTX group (62.0%) did not show radiographic progression (ΔmTSS ≤0.5) versus the MTX alone group (35.4%; p<0.001) (See Figure 2).
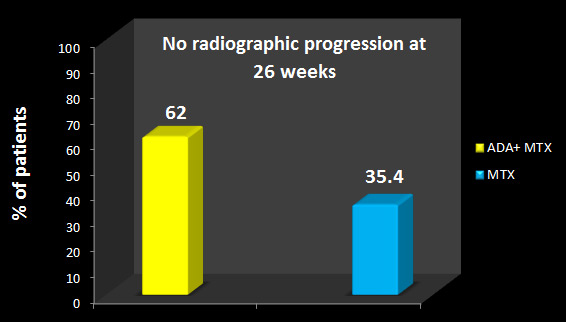
From above results it was concluded that Adalimumab in combination with low-dose MTX was efficacious in suppressing radiographic progression and improving clinical outcomes in Japanese patients with early RA and high disease activity.
A total of 87 patients were subjected to an evaluation of radiographic response to ADA after 52 weeks of treatment. The mean estimated yearly progression was 27.1 ± 46.0 at baseline, which is indicative of a great risk of further joint damage. After 52 weeks of ADA treatment, the mean change was significantly reduced to 0.8 ± 5.0.
ADA also suppressed the most aggressive progression in individuals with baseline changes of [100 TSS units/year].
The percentage of patients with no radiographic progression (as defined by a change in TSS of ≤0.5 units) over 52 weeks was 59.8%.
The results clearly indicate the ability of ADA to prevent further joint damage as assessed by a reduction in the rate of radiographic disease progression.
This trial was extension of DE0194 which evaluated the Radiographic Benefits of Longterm Adalimumab Plus Methotrexate over 10-years.
In DE019 trial, treatment arms were of ADA+ MTX and placebo (PBO) + MTX. They were given this treatment for 1 year and then patients from both the groups were allowed to receive treatment with ADA+ MTX for further 9 years. Results were assessed clinically, functionally and radiographically.
Radiographic nonprogression was defined as ∆mTSS ≤ 0.5 at Year 10. In addition, minimal radiographic progression was defined as ∆mTSS ≤ 5 at Year 10 (average of ≤ 0.5/yr, over 10 yrs)
Radiographic progression was minimal during this long term study, with a mean change from baseline to Year 10 in mTSS of 2.8 units (annual progression rate ~0.3 units/year).
44.8% had undetectable radiographic progression following up to 10 years of adalimumab + MTX therapy.
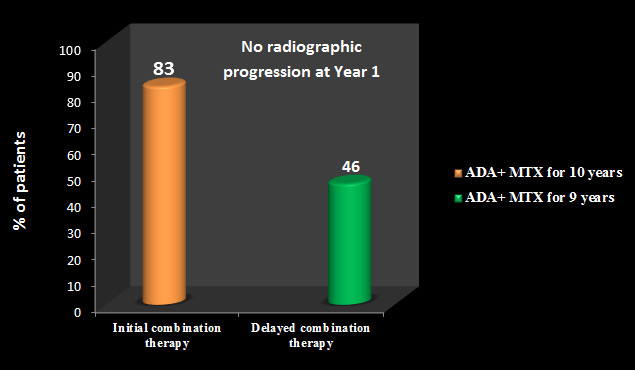
The percentages of patients with radiographic nonprogression (∆mTSS ≤ 0.5) at Year 1 were significantly greater for patients who received initial combination therapy (83% patients) compared with those who received delayed combination therapy (46% patients; p < 0.001) [See Figure 3].
[Note: Patients with initial combination therapy received ADA+ MTX for 10 years, whereas patients with delayed combination therapy received ADA+ MTX for 9 years].
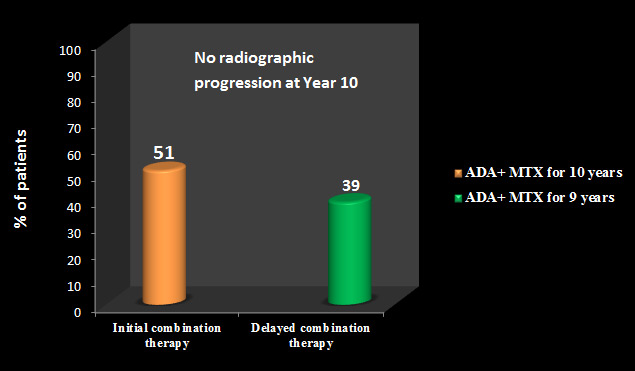
At Year 10, the percentage of patients with radiographic nonprogression was 51% for initial combination therapy vs. 39% for delayed combination therapy [See Figure 4].
Similar results were observed for the percentages of patients with minimal radiographic progression [∆mTSS ≤ 5 over 10 years: 77% vs. 65% for initial and delayed combination therapy groups, respectively].
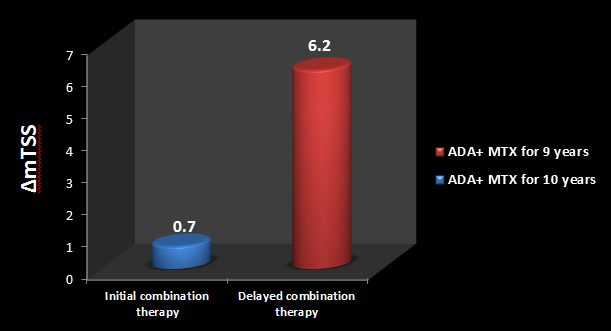
Still, the initial combination therapy group had significantly lower mean ∆mTSS compared with the delayed combination therapy group following 10 years of treatment [mean ∆mTSS (mTSS) = 0.7 vs.6.2; p = 0.002](See Figure 5)
Among patients who experienced radiographic progression ( ∆mTSS > 0.5) during the 10-year study, those who received initial combination therapy appeared to have less structural damage at Year 10 than those who received delayed combination therapy.
After 26 weeks, 87% of patients receiving combination therapy (ADA+ MTX) had no evidence of radiographic progression, compared with 72% of patients in the PBO+MTX group (p<0.001).
(See Figure 6)
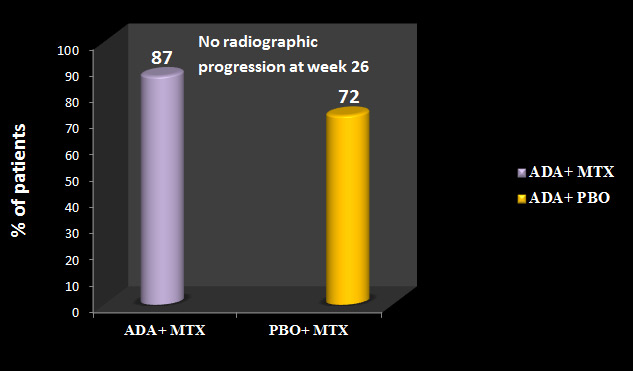
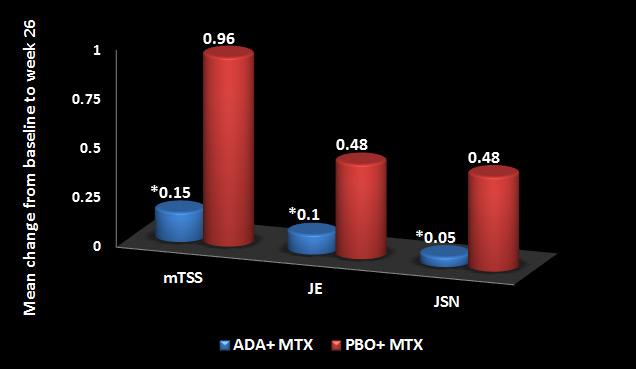
Figure 7 shown below demonstrates mean change in the van der Heijde modified total Sharp score (mTSS), joint erosion (JE) and joint space narrowing (JSN). *p<0.001
STAR Trial evaluated the safety and efficacy of Adalimumab , when given with standard antirheumatic therapy in patients with active rheumatoid arthritis (RA) not adequately responding to such therapies.
Similar rate of adverse events, serious adverse events, severe or life-threatening adverse events, and adverse events leading to withdrawal, as well as rates of infection and serious infection were seen between the 2 treatment groups. No cases of reactivated TB or opportunistic infections were reported.
Adalimumab Plus Standard |
Placebo Plus Standard Antirheumatic Therapy |
|
|---|---|---|
| Adverse events | 275 (86.5) | 263 (82.7) |
| Serious adverse events | 17 (5.3) | 22 (6.9) |
| Severe or life-threatening adverse events | 38 (11.9) | 49 (15.4) |
| Adverse events leading to withdrawal | 9 (2.8) | 7 (2.2 |
| Infection | 166 (52.2) | 157 (49.4) |
| Serious infection | 4 (1.3) | 6 (1.9) |
| Reactivated tuberculosis or opportunistic infections | 0 | 0 |
Rates of infections and serious infections did not vary by number of traditional DMARD used.
| 0 DMARD | 1 DMARD | |||
|---|---|---|---|---|
Adalimumab |
Placebo |
Adalimumab |
Placebo |
|
| Adverse events | 46 | 36 | 166 | 145 |
| Serious adverse events | 3 | 2 | 12 | 13 |
| Severe or life-threatening adverse events | 7 | 7 | 23 | 30 |
| Adverse events leading to withdrawal | 2 | 1 | 7 | 5 |
| Infection | 28 (49.1) | 17 (35.4) | 99 (53.8) | 93 (54.1) |
| Serious infection | 0 | 0 | 3 | 3 |
The most frequently reported (≥5%) adverse events that occurred in significantly greater proportions of adalimumab-treated patients were:
A total of 162 subjects were screened at 11 investigational sites in India, of which, 120 subjects were enrolled in the study, 60 subjects in each group viz. CHL’s Adalimumab and Innovator’s Adalimumab. Total 103 subjects were qualified as per protocol (PP) criteria and 119 subjects qualified for intend to treat (ITT) criteria which were included for efficacy analysis, respectively.
The American College of Rheumatology (ACR) response was significantly improved for each group at each evaluation throughout the study. After treatment with CHL’s Adalimumab, at Visit 5 (on day 84), 82% of patient had an ACR20, 46% had an ACR50 and 14% had an ACR70. Whereas in Innovator’s Adalimumab group at Visit 5 (on Day 84), 79.2% of patient had an ACR20, 43.4% had an ACR50 and 15.1% had an ACR70 response. No statistically significant difference between treatment groups were observed in ACR response.
In addition to swollen joints and tender joints, significant improvement was noted in all ACR core components. The difference in both treatment groups was not significant.
Significant improvement was observed in Disease Activity Score 28 C Reactive Protein (DAS28 CRP) score from the baseline in both the groups. The DAS28 CRP score in CHL’s Adalimumab group was 5.8 ± 0.88 and in Innovator’s Adalimumab group was 5.8 ± 0.83. Over a period of 84 days the score was decreased to 3.7 ± 1.12 and 3.7 ± 0.94 in CHL’s Adalimumab and Innovator’s Adalimumab group, respectively. No significant difference was observed between
treatment groups at baseline and end of the study.
Similarly, at baseline Disease Activity Score 28 Erythrocyte Sedimentation Rate (DAS28 ESR) score in CHL’s Adalimumab group was 6.8 ± 0.78 and in Innovator’s Adalimumab group it was 6.9 ± 0.81. Over a period of 84 days the score was decreased to 4.8 ± 1.04 and 4.8 ± 0.89 in CHL’s Adalimumab and Innovator’s Adalimumab group, respectively. No significant difference was observed between treatment groups at baseline and end of the study.
An efficacy subset analysis showed no significant difference between the treatment groups with respect to age groups, gender and weight on ACR criteria response and DAS 28 (CRP/ESR).
In this study, no significant difference was observed for anti-drug antibodies detected in samples from CHL’s Adalimumab treated and Innovator’s Adalimumab treated RA patients.
Anti-drug antibody was observed in two samples of patients treated with CHL’s Adalimumab on Visit 5 with titer value of 25 and 800, and one sample of patient treated with Innovator’s Adalimumab on Visit 5 with titre values of 200. These results indicate that both the drug products CHL’s Adalimumab and Innovator’s Adalimumab are similar with respect to immunogenic response in patients.
Visit |
Responder |
Adalimumab(CHL) |
Adalimumab (Innovator)(N = 53) |
|
|---|---|---|---|---|
| ACR20 | Visit 3 (Day 28) | Yes | 25 (50.0%) | 24(45.3%) |
| No | 25 (50.0%) | 29 (54.7%) | ||
| Visit 4 (Day 56) | Yes | 37 (74.0%) | 37 (69.8%) | |
| No | 13 (26.0%) | 16 (30.2%) | ||
| Visit 5 (Day 84) | Yes | 41 (82.0%) | 42 (79.2%) | |
| No | 9 (18.0%) | 11 (20.8%) | ||
| ACR50 | Visit 3 (Day 28) | Yes | 4 (8.0%) | 5 (9.4%) |
| No | 46 (92.0%) | 48 (90.6%) | ||
| Visit 4 (Day 56) | Yes | 17 (34.0%) | 10 (18.9%) | |
| No | 33 (66.0%) | 43 (81.1%) | ||
| Visit 5 (Day 84) | Yes | 23 (46.0%) | 23 (43.4%) | |
| No | 27 (54.0%) | 30 (56.6%) | ||
| ACR70 | Visit 3 (Day 28) | Yes | 1 (2.0%) | 3 (5.7%) |
| No | 49 (98.0%) | 50 (94.3%) | ||
| Visit 4 (Day 56) | Yes | 6 (12.0%) | 6 (11.3%) | |
| No | 44 (88.0%) | 47 (88.7%) | ||
| Visit 5 (Day 84) | Yes | 7 (14.0%) | 8 (15.1%) | |
| No | 43 (86.0%) | 45 (84.9%) |
Abbreviations: N = number of subjects in specified treatment; n = number of subjects at specified category.
ACR20, ACR50 and ACR70 responders: ≥20%, ≥50% and ≥70%, respectively, improvement in tender and swollen joint count; and ≥ 20%, ≥ 50% and ≥ 70%, respectively, improvement in at least 3 of 5 remaining ACR core measures: patient assessment of pain; patient and physician global assessment of disease activity; self-assessed disability [HAQ]; and CRP.
| ACR Core component | Adalimumab(CHL) (N=50) |
Adalimumab(Innovator) (N=53) |
|---|---|---|
| Tender joint count score (0-28) | ||
| Visit 1 (Day 1) | 16.6 ± 6.09 | 17.4 ± 6.32 |
| Change from baseline at Visit 5 (Day 84) |
-10.5 ± 5.95* | -11.7 ± 7.19* |
| Swollen joint count score | ||
| Visit 1 (Day 1) | 11.7 ± 5.57 | 12.4 ± 5.24 |
| Change from baseline atVisit 5 (Day 84) | -8.2 ± 5.77* | -9.2 ± 6.02* |
| Patient Assessment of pain | ||
| Visit 1 (Day 1) | 66.5 ± 12.38 | 66.4 ± 11.11 |
| Change from baseline at Visit 5 (Day 84) |
-30.0 ± 17.66* | -28.4 ± 16.75* |
| Patient global assessment of disease activity | ||
| Visit 1 (Day 1) | 66.2 ± 11.91 | 64.8 ± 10.57 |
| Change from baseline at Visit 5 (Day 84) |
-30.5 ± 16.75* | -28.3 ± 18.11* |
| Physician global assessment of disease activity | ||
| Visit 1 (Day 1) | 63.4 ± 12.02 | 63.9 ± 10.39 |
| Change from baseline at Visit 5 (Day 84) |
-29.2 ± 18.35* | -28.6 ± 18.02* |
| Disability Index of the HAQ | ||
| Visit 1 (Day 1) | 1.7 ± 0.62 | 1.6 ± 0.61 |
| Change from baseline at Visit 5 (Day 84) |
-0.8 ± 0.63* | -0.7 ± 0.60* |
| CRP | ||
| Visit 1 (Day 1) | 11.0 ± 12.72 | 10.5 ± 12.90 |
| Change from baseline at Visit 5 (Day 84) |
-5.5 ± 12.66* | -0.7 ± 26.98 |
| ESR | ||
| Visit 1 (Day 1) | 53.9 ± 21.45 | 53.2 ± 20.33 |
| Change from baseline at Visit 5 (Day 84) |
-8.6 ± 19.76* | -5.4 ± 17.35* |
| DAS 28-CRP | ||
| Visit 1 (Day 1) | 5.8 ± 0.88 | 5.8 ± 0.83 |
| Change from baseline at Visit 5 (Day 84) |
-2.1 ± 1.09* | -2.1 ± 1.21* |
| DAS 28-ESR | ||
| Visit 1 (Day 1) | 6.8 ± 0.78 | 6.9 ± 0.81 |
| Change from baseline at Visit 5 (Day 84) |
-2.0 ± 1.10* | -2.1 ± 1.15* |
Values presented in Mean ± SD.
* Significant compared to baseline.
Overall, CHL’s Adalimumab and Innovator’s Adalimumab were safe and well tolerated in this study. A total of 31 adverse events (AEs) including 3 serious adverse events (SAEs) were reported during the study, which were completely resolved. The distribution of AEs was comparable between the treatment groups. There were 15 AEs (including 2 SAEs) reported by 9 subjects in CHL’s Adalimumab treated group, whereas in Innovator’s Adalimumab treated group 16 AEs (including 1 SAE) were reported by 11 subjects. Pyrexia, headache and cough were commonly reported in both the treatment groups. The 3 SAEs reported were pyrexia, dizziness and cough. Majority of AEs were mild in intensity and not related to the study drugs. Three system organ class (SOC) with higher number of AEs were gastrointestinal disorder, general disorder and administration site condition, and infection and infestation.
Dyspnoea, Fungal infection, Gastritis, Headache, Injection site reaction , Joint swelling, Oligom enorrhoea, Pollakiuria, Polymenorrhoea, Pulmonary tuberculosis, Pyrexia, Rash, Urinary tract infection, Vomiting, Abdominal discomfort, Abdominal pain, Accelerated hypertension, Arthralgia, Asthenia, Body tinea, Chest pain, Cough, Diarrhoea and Dyspepsia.
There were no persistent changes from baseline in laboratory parameters in both treatment groups. General examination showed no significant sign in any treatment groups except 2 cases of pallor at visit 3 in Innovator’s Adalimumab treated group. No other systemic abnormality was observed throughout the study except musculoskeletal; however, the proportion of abnormality was comparable in both treatment groups.
The safety profile of CHL’s Adalimumab was assessed using a battery of toxicological studies.
Preclinical studies for CHL’s Adalimumab were performed as per the recommendations set forth in Schedule Y/ICH guidelines in compliance with GLP standards at Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad. Toxicological studies included independent acute toxicity studies in mice and rats by intended subcutaneous route and by alternate intravenous route of administration. Repeated dose toxicity studies by subcutaneous route comprising weekly dosing schedule over a period of four weeks was performed in rats and rabbits. Local tolerance evaluation was a part of repeated dose toxicity studies and by an independent skin sensitization study in guinea pigs.
In general, CHL’s Adalimumab revealed a good safety margin in terms of mortality over the acute dose of 830 mg/kg in mice & 410 mg/kg in rats by both subcutaneous and intravenous routes and were approximately 100 times of the human equivalent dose. No mortality, apparent signs of toxicity, adverse changes in body weights and gross pathological lesions were noticed in both mice and rats when compared to vehicle control groups. CHL’s Adalimumab did not induce any dermal sensitization in guinea pigs. No any adverse local tolerance effects were noticed at the site of injection in both rats and rabbits.
Repeated weekly subcutaneous administration of CHL’s Adalimumab was conducted over a period of four weeks at dose levels of 4.1, 20.5 & 41 mg/kg in rats and 2.1, 10.5 & 21 mg/kg in rabbits. The selected dose levels were 1X, 5X & 10X of the human equivalent dose. A recovery
group was maintained for a period of two weeks at 10X of the human equivalent dose. Vehicle control groups were maintained with main study & recovery groups. Innovator’s Adalimumab was used at 1X of the human equivalent dose for comparative purpose in repeated dose toxicity studies.
No mortality occurred in both rats and rabbits. No adverse changes were noticed during detailed clinical examination, body weight and feed intake determinations, hematological, biochemical, organ weight estimations, bone marrow examination and gross or histo-pathological evaluation. No differences were noticed in both the groups treated with CHL’s Adalimumab and Innovator’s Adalimumab in both rats and rabbits. No delayed toxicity was noticed during treatment free recovery period of two weeks. The immunogenic response in CHL’s Adalimumab treated groups was comparable to that of Innovator’s Adalimumab treated group. No immunogenicity titers were noticed against host cell proteins (HCP) which demonstrates that the product have extremely low levels of HCP contaminants.
NOAEL was considered to be more than 10X of human equivalent dose (41 mg/kg in rats and 21 mg/kg in rabbits) by weekly subcutaneous administration over a period of four weeks.
Thus, the overall pre-clinical profile of CHL’s Adalimumab seems to be non-inferior with the Innovator’s Adalimumab and considered to be safe at the recommended dose in humans.
The most commonly reported undesirable effects in patients taking Adalimumab include: infections (such as nasopharyngitis, upper respiratory tract infection and sinusitis), injection site reactions (erythema, itching, haemorrhage, pain or swelling), headache and musculoskeletal pain. Some fatal infections (including sepsis, opportunistic infections, TB), HBV reactivation and various malignancies (including leukaemia, lymphoma and HSTCL) have been reported. Serious reactions such as haematological, neurological and autoimmune reactions were also reported. Some rare reactions include pancytopenia, aplastic anaemia, central and peripheral
demyelinating events, lupus, lupus-related conditions and Stevens Johnson syndrome.
| System Organ Class | Frequency | Adverse Reaction |
|---|---|---|
| Infections and infestations | Very common | Respiratory tract infections (including lower and upper respiratory tract infection , pneumonia, sinusitis,pharyngitis, nasopharyngitis and pneumonia herpes viral) |
| Common | Systemic infections (including sepsis, candidiasis and influenza), intestinal infections (including gastroenteritis viral), skin and soft tissue infections (including paronychia, cellulitis, impetigo, necrotizing fasciitis and herpes zoster), ear infections, oralinfections (including herpes simplex, oral herpes and tooth infections), reproductive tract infections (including vulvovaginal mycotic infection), urinary tract infections ( including pyelonephritis), fungal infections |
|
| Uncommon | Neurological infections (including viral meningitis), opportunistic infections and tuberculosis ( includingcoccidioidomycosis, histoplasmosis and mycobacterium avum complex infection), bacterial infections, eye infections, joint infections, diverticulitis | |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | Common | Skin cancer excluding melanoma (including basal cell carcinoma and squamous cell carcinoma), benign neoplasm |
| Uncommon | Lymphoma, solid organ neoplasm (including breast cancer, lung neoplasm and thyroid neoplasm), melanoma | |
| Rare | Leukaemia | |
| Not known | hepatosplenic T-cell lymphoma | |
| Blood and the lymphatic system disorders |
Very common | leucopaenia (including neutropaenia and agranulocytosis), anaemia |
| Common | leucocytosis, thrombocytopaenia | |
| Uncommon | Idiopathic thrombocytopaenicpurpura | |
| Rare | Pancytopaenia | |
| Immune system disorders | Common | Hypersensitivity, allergies (including seasonal allergy) |
| Uncommon | Sarcoidosis | |
| Rare | Anaphylaxis | |
| Metabolism and nutrition disorders |
Very common | Lipids increased |
| Common | Hypokalaemia, uric acid increased, blood sodium abnormal, hypocalcaemia, hyperglycaemia, hypophosphataemia, dehydration | |
| Psychiatric disorders | Common | Mood alterations (including depression), anxiety, insomnia |
| Nervous system disorders | Very common | Headache |
| Common | Paraesthesias (including hypoaesthesia), migraine, sciatica | |
| Uncommon | Cerebrovascular accident , tremor, neuropathy | |
| Rare | Multiple sclerosis, demyelinating disorder (e.g. optic neuritis, Guillain-Barré syndrome) | |
| Eye disorders | Common | Visual impairment, conjunctivitis |
| Uncommon | Blepharitis, eye swelling, diplopia | |
| Ear and labyrinthdisorders | Common | Vertigo |
| Uncommon | Deafness, tinnitus | |
| Cardiac disorders | Common | Tachycardia |
| Uncommon | Myocardial infarction, arrhythmia, congestive heart failure | |
| Rare | Cardiac arrest | |
| Vascular disorders | Common | Hypertension, flushing, haematoma |
| Uncommon | Aortic aneurysm, vascular arterial occlusion, thrombophlebitis | |
| Respiratory, thoracic and mediastinal disorders |
Common | Asthma, dyspnoea, cough |
| Uncommon | pulmonary embolism, interstitial lung disease, chronic obstructive pulmonary disease, pneumonitis pleural effusion | |
| Rare | Pulmonary fibrosis | |
| Gastrointestinal disorders | Very common | Abdominal pain, nausea and vomiting |
| Common | GI haemorrhage, dyspepsia, gastroesophageal reflux disease, sicca syndrome | |
| Uncommon | Pancreatitis, dysphagia, face oedema | |
| Rare | Intestinal perforation | |
| Hepato-biliary disorders | Very common | Elevated liver enzymes |
| Uncommon | Cholecystitis and cholelithiasis, hepatic steatosis, bilirubin increased | |
| Rare | Reactivation of hepatitis B | |
| Skin and subcutaneous tissue disorders |
Very common | Rash (including exfoliative rash) |
| Common | Worsening or new onset of psoriasus (including palmplantarpustular psoriasis), urticaria, bruising (including purpura), dermatitis (including eczema), onychoclasis, hyperhydrosis, alopecia, pruritus | |
| Uncommon | Night sweats, scar | |
| Rare | Erythema multiforme, Stevens-Johnson syndrome, angioedema,cutaneous vasculitis | |
| Musculoskeletal, connective tissue and bone disorders |
Very common | Musculoskeletal pain |
| Common | Muscle spasms (including blood creatine phosphokinase increased) | |
| Uncommon | Rhabdomyolysis, systemic lupus erythematosus | |
| Rare | Lupus-like syndrome | |
| Renal and urinary disorders |
Common | Renal impairment, haematuria |
| Uncommon | Nocturia | |
| Reproductive system and breast disorders |
Uncommon | Erectile dysfunction |
| General disorders and administration site conditions |
Very common | Injection site reaction (including injection site erythema) |
| Common | Chest pain, oedema | |
| Uncommon | Inflammation | |
| Investigations | Common | Coagulation and bleeding disorders (including activated partial thromboplastin time prolonged), autoantibody test positive (including double stranded DNA antibody), blood lactate dehydrogenase increased |
| Injury and poisoning | Common | Impaired healing |